India has the second highest burden of sickle cell disease in the world.
Speaking at an Apollo Hospitals event in June, Dr Gaurav Kharya, clinical lead at the Centre for Bone Marrow Transplant and Cellular Therapy, identified a “‘sickle cell belt’, that is, a significant population of the Deccan plateau of Central India, northern Kerala and Tamil Nadu.” He also noted that the disease is most prevalent in southern Indian states, including Chhattisgarh, Bihar, Maharashtra and neighboring areas of Madhya Pradesh, although the disease is still widespread across India. Cases are most often found among rural and tribal communities.
Genetic background and symptoms
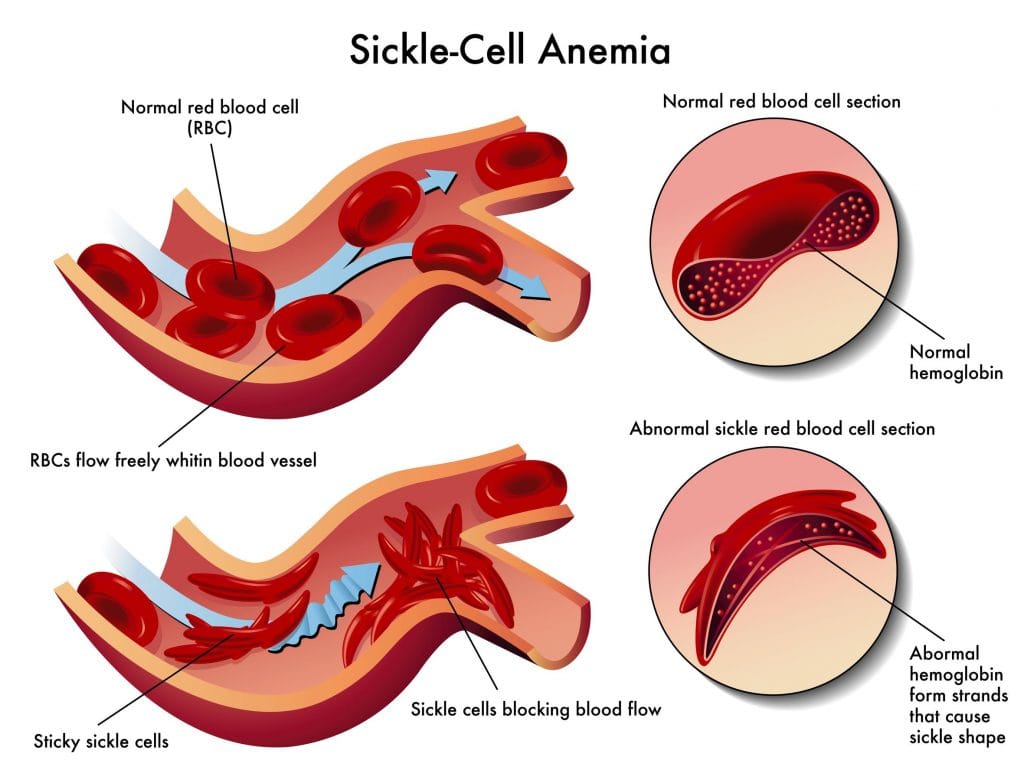
Sickle cell disease disease is an inherited genetic condition that affects a single gene. Abnormal alleles of the gene cause defective copies of the haemoglobin protein — responsible for the transport of oxygen — to be formed.
Heterozygous individuals — or individuals with only a single allele of the defective gene — are referred to as carriers. These individuals may have very mild symptoms, or be entirely asymptomatic. As such, two individuals may opt to have children without knowing whether they possess abnormal genes. In these situations a child has the potential to have two copies of the abnormal gene. The child will then have full, symptomatic sickle cell disease.
The defective haemoglobin can lead to red blood cells taking up a sickle-like shape, rather than their typical round shape. Issues can arise when these sickle-shaped cells interconnect with other malformed red blood cells. This can lead to blood clotting, a condition which can result in symptoms such as severe pain, but can also cause life threatening complications such as strokes and organ failure.
Prevalence of sickle cell disease
One theory on why the disease is so abundant — termed the “malaria hypothesis” — states that the prevalence in Africa, as well as other regions in areas such as rural India, may be far higher due to a selective effect for individuals with a single sickle cell gene. These studies have assessed infant mortality rates in high prevalence regions and found slight decreases in deaths. This, it was found, is due to a protective effect the gene confers against the effects of malaria.
The mechanism by which this protection occurs is the structural change to red blood cells. This structural change in which healthy, rounded red blood cells are altered to the sickle-like form causes the cells to rupture far earlier in the malaria parasite’s breeding cycle, thereby reducing the capacity of malaria to spread within an individual. This limits the effects of symptoms such as malarial anaemia within SCD carrier individuals.
This protective effect against malaria symptoms has made heterozygous carrier individuals better able to survive in areas where malaria is endemic, such as India and Africa. This greater ability to survive in these regions is offset by the higher mortality rate of homozygous individuals with symptomatic sickle cell disease.
However, genetic modelling shows the heterozygous trait has a greater chance of being passed to the next generation from two carrier parents than homozygous sickle cell disease. As a single gene trait, two heterozygous individuals have a 25 percent chance of having a perfectly healthy child; a 25 percent chance of a homozygous sickle cell disease-affected child; and a fifty percent chance of a child who is also a carrier.
Therefore, in a malaria-endemic region, a protective effect would be conferred on a greater portion of the population than would be affected by full-symptomatic sickle cell disease — creating a situation in which heterozygotes are better able to survive and pass on the genes.
Prevention and treatment
Dr Kharya said that knowing one’s disease status is the most important factor in addressing the condition. Those who are homozygous for the condition will likely be aware of their disease status through persistent symptoms such as anaemia or pain related to blood clotting. Some, however, may still remain undiagnosed through lack of access to health infrastructure.
For those who are carriers, their symptoms may be entirely absent. In these cases, the individual may never have had a blood test and so remain unaware of their potential to pass on sickle cell disease to their children.
Awareness of disease status for carriers is imperative for family planning. If one parent is aware that they are a carrier, they may opt to have children with an individual who is confirmed to not have the disease, as this will produce the greatest chance of having children who do not possess sickle cell genes.
Diagnosis at an early stage will also allow for reduction of mortality through access to correct treatment. Lack of knowledge of the disease means an individual may not seek out treatment, in turn increasing the risk of blood clots leading to potential death.
The disease can potentially be addressed through bone marrow donation, though the procedure can cause complications, even death. Donor matches are rare and so this treatment is not standard procedure. As a result, a focus is placed on symptom management, largely revolving around the management of pain resulting from blood clots.